

The Role of CFTR Mutations in Causing CF
Cystic fibrosis (CF) is a complex genetic disease characterized by a functional disorder of the exocrine glands, which can affect lung function, digestion, sweat production, and reproductive function.1 The disease occurs when a child inherits two copies of a cystic fibrosis transmembrane conductance regulator (CFTR) gene mutation, resulting in a CF genotype.1 CF phenotypes can show variability, even among patients with the same mutations.2
What Do CFTR Proteins Do?
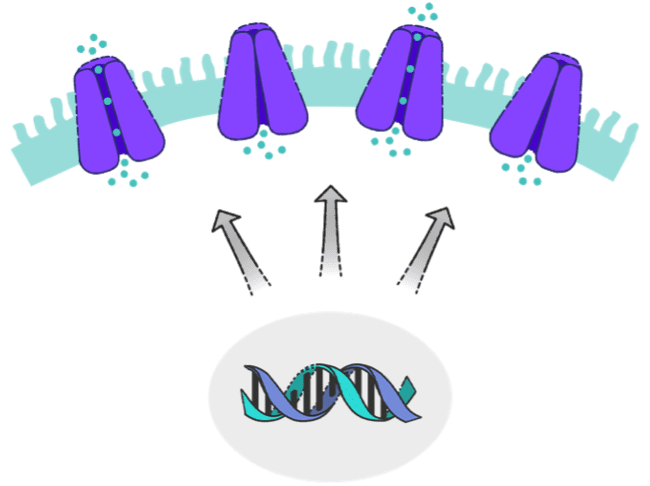
CFTR proteins are found on the surfaces of epithelial cells in various organs in the body.2,3 Normally, CFTR protein channels transport ions, such as chloride and bicarbonate, into and out of epithelial cells in these organs.1
CFTR protein activity is largely determined by the quantity and function of the protein.1,3 Different CFTR mutations affect protein quantity and function in different ways.1,4
When there is an imbalance of salt and water transport (ionic transportation), the result can be thick, viscous mucus.1,3 This leads to infections, inflammation, tissue damage, and other phenotypic manifestations common to CF1,3:
- Characteristically, CFTR mutations may cause problems that affect the reabsorption of chloride and sodium, and as a result, cause the sweat glands to secrete excessive amounts of sodium chloride.1
- In the pancreas and gastrointestinal tract, the defective secretion of digestive enzymes and malabsorption of fat can lead to fatty stools.1 85% to 90% of patients with CF have pancreatic insufficiency, which is common even at a young age.5
- CF can significantly impact the lung airways. If left untreated, mucus clogging of small airways can lead to repeated bacterial infection, inflammation, bronchiectasis, and ultimately an accelerated decline in lung function.1
- CF also affects reproductive fertility, particularly in males. Approximately 98% of males with CF have a congenital bilateral absence of the vas deferens, blocking the transport of sperm and resulting in azoospermia.6 Infertility in women with CF is not as common, with up to 50% of females able to conceive.6
Maintaining good water and salt balance at the epithelial cell surface requires both adequate quantity and adequate function of CFTR proteins. If either of these is compromised, CF can result.7




How is CF Inherited?
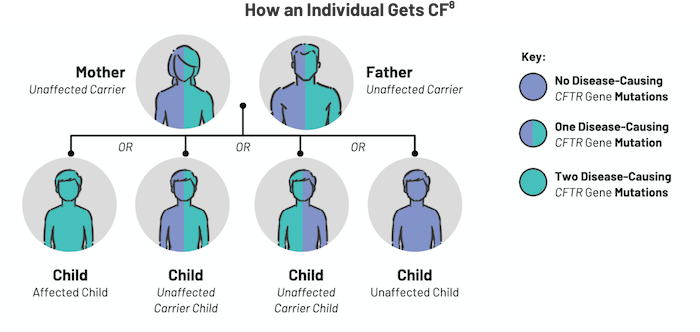
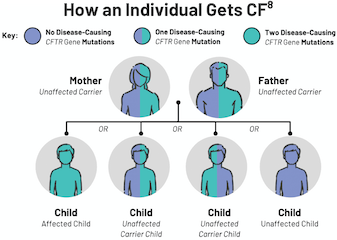
CF is inherited in an autosomal recessive pattern.3 In order to have CF, an individual must inherit two mutated copies of the CFTR gene, which leads to a CFTR protein dysfunction.3 CFTR protein dysfunction impairs cellular chloride and bicarbonate transport, resulting in the clinical manifestation of CF.1,4
Conversation Considerations for Caregivers
Conversation Considerations for Caregivers
Conversation Considerations
for Caregivers
Below are some common questions a patient or caregiver may ask when learning about CF, with suggested responses in patient-friendly language that may help them more easily understand.⁹ These questions are provided as examples.
What are genes and how do they relate to CF?
- Genes are the instruction manual of the body. Sometimes there will be a mistake, or a mutation, in the genes which can result in a disease.10 When it comes to CF, there’s a mutation in the CFTR gene.8
What’s the job of CFTR proteins?
- When a CFTR protein is working correctly, it forms a channel so that salt and water are able to move freely through cells, allowing the cells that produce the following to work properly8:
- Mucus
- Sweat
- Digestive enzymes
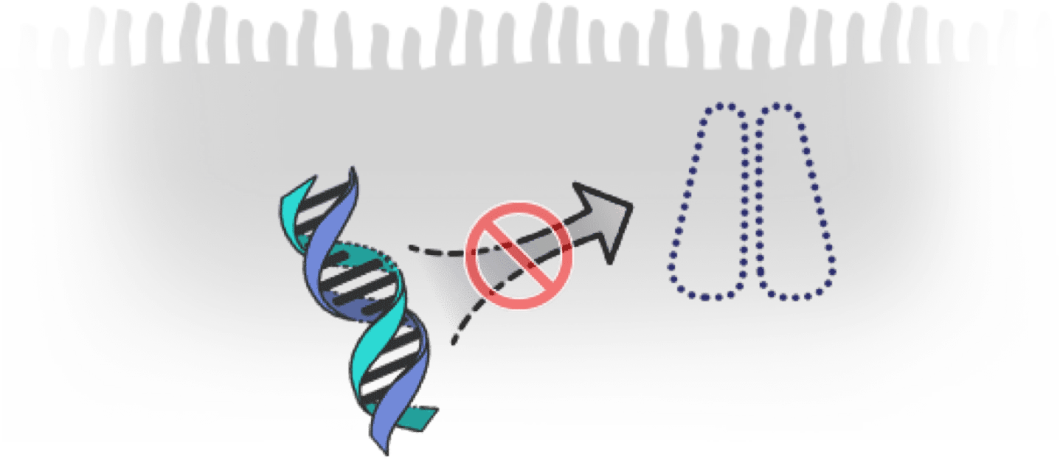
How can genetic mutations affect CFTR proteins?
- Genetic mutations can affect CFTR proteins in different ways, resulting in problems including13:
- CFTR proteins not working effectively
- No CFTR proteins produced
CFTR proteins not working effectively
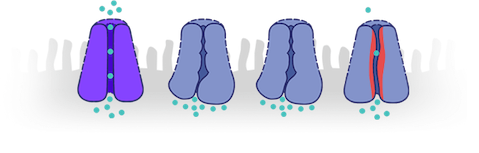
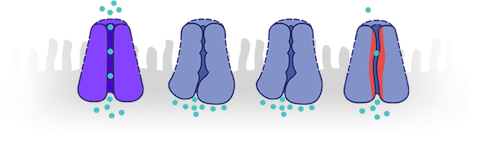
No CFTR proteins produced

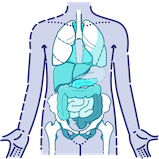
What parts of the body are affected by CF?
Having basic visuals like the one below may help communicate your message with your patients and their caregivers9,12:






You can also direct your patients and their caregivers to CFSource.com for comprehensive videos and downloadable guides that may help them learn more about a variety of CF topics
You May Also Be Interested In
References: 1. Derichs N. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev. 2013;22(127):58-65. doi:10.1183/09059180.00008412 2. Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67(2):117‐133. doi:10.1159/000029497 3. Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med. 2013;1(2):158-163. doi:10.1016/S2213-2600(12)70057-7 4. Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519-2531. doi:10.1016/S0140-6736(16)00576-6 5. Orenstein DM, Spahr JE, Weiner DJ. Cystic fibrosis: a guide for patient and family. 4th ed. Lippincott Williams & Wilkins; 2011. 6. Ahmad A, Ahmed A, Patrizio P. Cystic fibrosis and fertility. Curr Opin Obstet Gynecol. 2013;25(3):167-172. doi:10.1097/GCO.0b013e32835f1745 7. Feng LB, Grosse SD, Green RF, Fink AK, Sawicki GS. Precision medicine in action: the impact of ivacaftor on cystic fibrosis–related hospitalizations. Health Aff (Millwood). 2018;37(5):773-779. doi:10.1377/hlthaff.2017.1554 8. Cunningham JC, Taussig LM. An introduction to cystic fibrosis for patients and their families. Cystic Fibrosis Foundation. Accessed June 22, 2023. https://www.cff.org/sites/default/files/2021-09/Intro-to-CF.pdf 9. Borowitz D, Robinson KA, Rosenfeld M, et al. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155(suppl 6):S73-93. doi:10.1016/j.jpeds.2009.09.001 10. How can gene mutations affect health and development? U.S. National Library of Medicine. MedlinePlus website. Updated March 25, 2021. Accessed June 22, 2023. https://medlineplus.gov/genetics/understanding/mutationsanddisorders/mutationscausedisease/ 11. MacDonald KD, McKenzie KR, Zeitlin PL. Cystic fibrosis transmembrane regulator protein mutations: ‘class’ opportunity for novel drug innovation. Paediatr Drugs. 2007;9(1):1-10. doi:10.2165/00148581-200709010-00001 12. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373 (9678):1891-1904. doi:10.1016/S0140-6736(09)60327-5 13. Amaral MD. Novel personalized therapies for cystic fibrosis: treating the basic defect in all patients. J Intern Med. 2015;277(2):155-166. doi:10.1111/joim.12314