

Multi-Organ Disease Progression
Cystic fibrosis (CF) is a progressive and multisystemic disease that can affect any organ that exhibits significant exocrine function.1,2 CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene; these CFTR abnormalities can cause a domino effect, leading to complications in several organs.2–4
Effects of CFTR Protein Dysfunction in the Lungs
CFTR protein abnormalities result in a domino effect, which ultimately leads to structural damage in several organs. For example, in the lungs3–6:
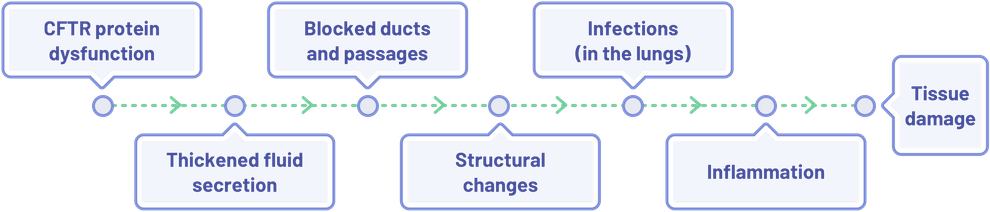
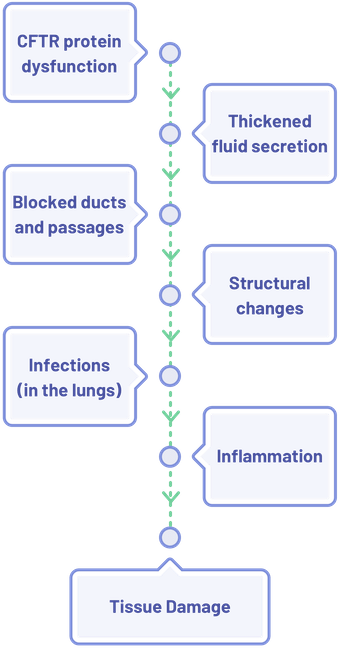
A similar cascade of effects occurs in other organ systems as well; with the end result being inflammation leading to organ and tissue damage.1,3,4,6–12
For many individuals with CF, symptoms manifest early in life, with certain signs appearing in utero.3,8–10,13 Additionally, certain organ damage, including: lungs, liver, or pancreas, can occur before symptoms.1,10,14 As individuals with CF age, complications from CF extend beyond lung disease.1,15–17 As a result, patients with CF require complete care from multidisciplinary teams as their complications change over time.1,15–17
For many individuals with CF, symptoms manifest early in life, with certain signs appearing in utero.3,8–10,13 Additionally, certain organ damage, including: lungs, liver, or pancreas, can occur before symptoms.1,10,14 As individuals with CF age, complications from CF extend beyond lung disease. 1,15–17 As a result, patients with CF require complete care from multidisciplinary teams as their complications change over time.1,15–17
For many individuals with CF, symptoms manifest early in life, with certain signs appearing in utero.3,8–10,13 Additionally, certain organ damage, including: lungs, liver, or pancreas, can occur before symptoms.1,10,14 As individuals with CF age, complications from CF extend beyond lung disease.1,15–17 As a result, patients with CF require complete care from multidisciplinary teams as their complications change over time.1,15–17
Disease Progression in the Lungs
Disease Progression in the Lungs
Disease Progression in the Lungs
In a patient with CF, lung disease begins early and develops throughout their lifetime.3
| Lung Disease Progression3,10,13,17 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Early mucinous |
Advancing bronchiectasis and pulmonary exacerbations
 |
Bronchiectasis with recurrent exacerbations, requiring nutritional support, supplementary oxygen, and noninvasive ventilator support
 |
Bronchiectasis with hemoptysis, pneumothorax;  |
| Additional Considerations |
Potentially irreversible damage as early as 2 years of age; eventually pulmonary insufficiency is responsible for ~80% of CF-related deaths |
||
| Lung Disease Progression3,10,13,17 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Early mucinous |
Advancing bronchiectasis and pulmonary exacerbations
|
Bronchiectasis with recurrent exacerbations, requiring nutritional support, supplementary oxygen, and noninvasive ventilator support
|
Bronchiectasis with hemoptysis, pneumothorax; |
| Additional Considerations |
Potentially irreversible damage as early as 2 years of age; eventually pulmonary insufficiency is responsible for ~80% of CF-related deaths |
||
| Lung Disease Progression3,10,13,17 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Early mucinous |
Advancing bronchiectasis and pulmonary exacerbations
|
Bronchiectasis with recurrent exacerbations, requiring nutritional support, supplementary oxygen, and noninvasive ventilator support
|
Bronchiectasis with hemoptysis, pneumothorax; |
| Additional Considerations |
Potentially irreversible damage as early as 2 years of age; eventually pulmonary insufficiency is responsible for ~80% of CF-related deaths |
||
Disease Progression in the Pancreas
CF can affect both the exocrine and endocrine functions, which can result in pancreatic insufficiency.9,10

Exocrine
Exocrine
Exocrine
Clogged pancreatic ducts prevent digestive enzymes from passing into the intestines, causing the pancreas to break down9,10
Endocrine
Endocrine
Endocrine
CFTR protein dysfunction can result in impaired insulin secretion and carbohydrate intolerance, leading to cystic fibrosis-related diabetes mellitus (CFRD)9,10
Pancreatic insufficiency can begin at birth and progresses throughout a patient’s life.3,4
| Pancreatic Insufficiency Progression3,9,10,13,19 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Pancreatic exocrine insufficiency; up to 71% of patients with CF are pancreatic insufficient at birth
|
Up to 2% of patients <10 years of age may have CF-related diabetes mellitus  |
Up to 19% of adolescents have CF-related diabetes mellitus  |
Up to 40%-50% of adults have CF-related diabetes mellitus  |
| Additional Considerations |
Nutritional/caloric deficiency issue (growth impairment) |
||
| Pancreatic Insufficiency Progression3,9,10,13,19 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Pancreatic exocrine insufficiency; up to 71% of patients with CF are pancreatic insufficient at birth
|
Up to 2% of patients <10 years of age may have CF-related diabetes mellitus |
Up to 19% of adolescents have CF-related diabetes mellitus |
Up to 40%-50% of adults have CF-related diabetes mellitus |
| Additional Considerations |
Nutritional/caloric deficiency issue (growth impairment) |
||
| Pancreatic Insufficiency Progression3,9,10,13,19 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Pancreatic exocrine insufficiency; up to 71% of patients with CF are pancreatic insufficient at birth
|
Up to 2% of patients <10 years of age may have CF-related diabetes mellitus |
Up to 19% of adolescents have CF-related diabetes mellitus |
Up to 40%-50% of adults have CF-related diabetes mellitus |
| Additional Considerations |
Nutritional/caloric deficiency issue (growth impairment) |
||
Disease Progression in the Liver
CFTR protein dysfunctions can cause altered liver secretions, which can result in changed bile viscosity, causing the blockage of bile ducts.1,10,11 Subsequently, this blockage can result in liver scarring, inflammation, abnormal liver function, and eventually, liver damage.1,11,12
| Liver Damage Progression3,10,20 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Onset of abnormal liver function tests  |
5% of patients may
develop biliary cirrhosis by age 15 years  |
5%-10% of patients develop portal hypertension and a liver transplant may be required in some patients (typically age >35 years) |
|
| Liver Damage Progression3,10,20 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Onset of abnormal liver function tests |
5% of patients may
develop biliary cirrhosis by age 15 years |
5%-10% of patients develop portal hypertension and a liver transplant may be required in some patients (typically age >35 years) |
|
| Liver Damage Progression3,10,20 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
Onset of abnormal liver function tests | 5% of patients may develop biliary cirrhosis by age 15 years | 5%-10% of patients develop portal hypertension and a liver transplant may be required in some patients (typically age >35 years)  | |
Disease Progression in the Gastrointestinal Tract
Gastrointestinal complications and symptoms can occur throughout a patient with CF’s lifetime.3,10,11,13,17,20–22
| Gastrointestinal Complications and Symptoms3,10,11,13,17,20–22 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Up to 21% of newborns with CF have gastrointestinal problems, such as meconium ileus, within the first days of life |
Constant vigilance regarding malnutrition  |
Decreased motility may lead to chronic constipation  |
Distal intestinal obstruction syndrome (DIOS)  |
| Additional Considerations |
Gastrointestinal abnormalities are often evident before birth |
||
| Gastrointestinal Complications and Symptoms3,10,11,13,17,20–22 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Up to 21% of newborns with CF have gastrointestinal problems, such as meconium ileus, within the first days of life |
Constant vigilance regarding malnutrition |
Decreased motility may lead to chronic constipation |
Distal intestinal obstruction syndrome (DIOS) |
| Additional Considerations |
Gastrointestinal abnormalities are often evident before birth |
||
| Gastrointestinal Complications and Symptoms3,10,11,13,17,20–22 | |||
|---|---|---|---|
| 0-5 years | 6-10 years | 11-20 years | 20+ years |
|
Up to 21% of newborns with CF have gastrointestinal problems, such as meconium ileus, within the first days of life |
Constant vigilance regarding malnutrition |
Decreased motility may lead to chronic constipation |
Distal intestinal obstruction syndrome (DIOS) |
| Additional Considerations |
Gastrointestinal abnormalities are often evident before birth |
||
Progression of Bone Disease
Due to increasing evidence, it is suggested that CFTR dysfunction affects bone metabolism.23 The prevalence of bone disease increases in older patient populations.17,24 Between ages 20-35, a patient with CF may demonstrate arthropathy and CF-related bone disease; these patients are also at an increased risk of low bone density fractures.3,24
Bone Disease Progression17
<18 years
- 1.0% osteopenia
- 0.3% osteoporosis
≥18 years
- 18.0% osteopenia
- 7.5% osteoporosis
Disease Progression of Reproductive Organs
CFTR gene mutations may impair normal development of both male and female reproductive tracts, and as a result, CF can lead to infertility in both men and women.²
Reproductive Organs Disease Progression2

Male Infertility
- Mutations in the CFTR gene can cause congenital bilateral absence of the vas deferens (CBAVD)
- 98% of males with CF are infertile with obstructive azoospermia
- Mutations in the CFTR gene can cause congenital bilateral absence of the vas deferens (CBAVD)
- 98% of males with CF are infertile with obstructive azoospermia
- Mutations in the CFTR
gene can cause congenital bilateral absence of the vas deferens (CBAVD) - 98% of males with CF are infertile with obstructive azoospermia

Female Infertility
- Women can experience fertility impairment related to thick cervical mucus that fails to undergo the usual mid-cycle thinning
The Importance of Monitoring Multi-Organ Disease Progression
As advances in CF knowledge and care are potentially able to prolong the life expectancy of many patients with CF, it’s important to keep in mind the complications—beyond lung disease—that will develop and progress as patients age.1,15-17 Monitoring for these complications can help detect their emergence and progression, which can ensure earlier intervention; this has been associated with better outcomes in patients.1,13,14,16,17,20,25
Conversation Considerations for Caregivers
Conversation Considerations for Caregivers
Conversation Considerations
for Caregivers
You may want to discuss with your patients and caregivers how CF affects different organs in different ways, as this may help them be more proactive in the decision-making process and help them make more effective decisions in collaboration with their CF care team.3,4,13,14 These discussion tips are provided as suggestions.
You may want to discuss with your patients and caregivers how CF affects different organs in different ways, as this may help them be more proactive in the decision-making process and help them make more effective decisions in collaboration with their CF care team.3,4,13,14 These discussion tips are provided as suggestions.
You may want to discuss with your patients and caregivers how CF affects different organs in different ways, as this may help them be more proactive in the decision-making process and help them make more effective decisions in collaboration with their CF care team.3,4,13,14 These discussion tips are provided as suggestions.
For a guide that could be helpful in aiding conversations about how CF can progress, visit CFSource.com
ppFEV1, percent predicted forced expiratory volume in 1 second.
You May Also Be Interested In

References: 1. Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol. 2014;9(3):136-141. doi:10.5114/pg.2014.43574 2. Ahmad A, Ahmed A, Patrizio P. Cystic fibrosis and fertility. Curr Opin Obstet Gynecol. 2013;25(3):167-172. doi:10.1097/GCO.0b013e32835f1745 3. Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519-2531. doi:10.1016/S0140-6736(16)00576-6 4. Rana M, Munns CF, Selvadurai H, Donaghue KC, Craig ME. Cystic fibrosis-related diabetes in children—gaps in the evidence? Nat Rev Endocrinol. 2010;6(7):371-378. doi:10.1038/nrendo.2010.85 5. Cantin AM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros. 2015;14(4):419-430. doi:10.1016/j.jcf.2015.03.003 6. Sathe MN, Freeman AJ. Gastrointestinal, pancreatic, and hepatobiliary manifestations of cystic fibrosis. Pediatr Clin N Am. 2016;63(4):679-698. doi:10.1016/j.pcl.2016.04.008 7. Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173(5):475-482. doi:10.1164/rccm.200505-840OE 8. Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67(2):117‐133. doi:10.1159/000029497 9. Gibson-Corely KN, Meyerholz DK, Engelhardt JF. Pancreatic pathophysiology in cystic fibrosis. J Pathol. 2016;238(2):311-320. doi:10.1002/path.4634 10. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891-1904. doi:10.1016/S0140-6736(09)60327-5 11. Averill S, Lubner MG, Menias CO, et al. Multisystem imaging findings of cystic fibrosis in adults: recognizing typical and atypical patterns of disease. AJR Am J Roentgenol. 2017;209(1):3-18. doi:10.2214/AJR.16.17462 12. Cañas T, Maciá A, Muñoz-Codoceo RA, et al. Hepatic and splenic acoustic radiation force impulse shear wave velocity elastography in children with liver disease associated with cystic fibrosis. Biomed Res Int. 2015;2015:517369. doi:10.1155/2015/517369 13. VanDevanter DR, Kahle JS, O’Sullivan AK, Sikirica S, Hodgkins PS. Cystic fibrosis in young children: a review of disease manifestation, progression, and response to early treatment. J Cyst Fibros. 2016;15(2):147-157. doi:10.1016/j.jcf.2015.09.008 14. Ellemunter H, Fuchs SI, Unsinn KM, et al. Sensitivity of lung clearance index and chest computed tomography in early CF lung disease. Respir Med. 2010;104(12):1834-1842. doi:10.1016/j.rmed.2010.06.010 15. Naehrig S, Chao CM, Naehrlich L. Cystic fibrosis: diagnosis and treatment. Dtsch Arztebl Int. 2017;114(33-34):564-574. doi:10.3238/arztebl.2017.0564 16. Ronan NJ, Elborn JS, Plant BJ. Current and emerging comorbidities in cystic fibrosis. Presse Med. 2017;46(6):e125-e138. doi:10.1016/j.lpm.2017.05.011 17. Cystic Fibrosis Foundation. 2021 patient registry annual data report. Accessed June 27, 2023. https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf 18. de Jong PA, Ottink MD, Robben SG, et al. Pulmonary disease assessment in cystic fibrosis: comparison of CT scoring systems and value of bronchial and arterial dimension measurements. Radiology. 2004;231(2):434-439. doi:10.1148/radiol.2312021393 19. Lahiri T, Hempstead SE, Brady C, et al. Clinical practice guidelines from the Cystic Fibrosis Foundation for preschoolers with cystic fibrosis. Pediatrics. 2016;137(4):e20151784. doi: 10.1542/peds.2015-1784 20. Gelfond D, Borowitz D. Gastrointestinal complications of cystic fibrosis. Clin Gastroenterol Hepatol. 2013;11(4):333-342. doi:10.1016/j.cgh.2012.11.006 21. Lavie M, Manovitz T, Vilozni D, et al. Long-term follow-up of distal intestinal obstruction syndrome in cystic fibrosis. World J Gastroenterol. 2015;21(1):318-325. doi:10.3748/wjg.v21.i1.318 22. Abraham JM, Taylor CJ. Cystic fibrosis & disorders of the large intestine: DIOS, constipation, and colorectal cancer. J Cyst Fibros. 2017;16(suppl 2):S40-S49. doi:10.1016/j.jcf.2017.06.013 23. Marquette M, Haworth CS. Bone health and disease in cystic fibrosis. Paediatr Respir Rev. 2016;20(suppl):2-5. doi:10.1016/j.prrv.2016.06.003 24. Jacquot J, Delion M, Gangloff S, Braux J, Velard F. Bone disease in cystic fibrosis: new pathogenic insights opening novel therapies. Osteoporos Int. 2016;27(4):1401-1412. doi:10.1007/s00198-015-3343-3 25. Chedevergne F, Sermet-Gaudelus I. Prevention of osteoporosis in cystic fibrosis. Curr Opin Pulm Med. 2019;25(6):660-665. doi:10.1097/MCP.0000000000000624 26. Foil KE, Powers A, Raraigh KS, Wallis K, Southern KW, Salinas D. The increasing challenge of genetic counseling for cystic fibrosis. J Cyst Fibros. 2019;18(2):167-174. doi:10.1016/j.jcf.2018.11.014